1. Giai đoạn trước xét nghiệm
1.1. Các
yêu cầu của giai đoạn trước xét nghiệm
Người làm xét nghiệm cần kiểm tra phiếu yêu cầu xét nghiệm về các thông tin
liên quan đến bệnh nhân và yêu cầu xét nghiệm; Nhập sổ nhận mẫu và ghi nhận các
thông tin về cách thu thập, bảo quản, vận chuyển mẫu bệnh phẩm.
1.2. Quá
trình cần thực hiện trước xét nghiệm
1.2.1. Thông tin cho người yêu cầu xét
nghiệm
Thông tin cho người yêu cầu xét nghiệm bao gồm: Tên thường dùng và địa chỉ
của phòng xét nghiệm; Các thành viên chủ chốt của đơn vị chịu trách nhiệm; Địa
chỉ của phòng xét nghiệm; Thời gian làm việc của phòng xét nghiệm; Hướng dẫn về
việc vận chuyển mẫu, bao gồm cả việc đóng gói với mẫu bệnh phẩm đặc biệt; Có
thể có tư vấn và giải thích về lâm sàng; Các giới hạn về mặt thời gian trả lời
kết quả xét nghiệm và các yêu cầu xét nghiệm bổ sung.
1.2.2. Phiếu yêu cầu xét nghiệm
Phiếu yêu cầu xét nghiệm được thiết kế chính xác và được điền đầy đủ các
thông tin cần thiết để giúp cho các cán bộ xét nghiệm có thể hoàn thành nhiệm
vụ một cách thuận lợi. Các thông tin cần thiết bao gồm: Thông tin cá nhân và
nơi ở của bệnh nhân; Thời gian lấy mẫu; Loại mẫu và vị trí lấy mẫu trong cơ
thể; Yêu cầu xét nghiệm; Thời gian phòng xét nghiệm tiếp nhận mẫu; Các thông
tin lâm sàng liên quan; Nơi kết quả xét nghiệm được gửi tới,...
1.2.3. Lấy mẫu và bảo quản mẫu
Chuẩn bị bệnh nhân, lấy mẫu và bảo quản mẫu một cách phù hợp là điều kiện
cần thiết để thu được những kết quả xét nghiệm có giá trị. Phòng xét nghiệm cần
thực hiện các quy định về lấy mẫu và bảo quản mẫu, bao gồm: Việc hoàn thành
phiếu yêu cầu và khẳng định các thông tin về
bệnh nhân; Việc đánh dấu mẫu chính xác hay không? Kiểm tra xem bệnh nhân
đã được chuẩn bị đúng hay không? Xác định chắc chắn rằng đã lấy mẫu xét nghiệm
một cách chính xác; Hạn chế các nguy cơ làm biến đổi mẫu trong quá trình lấy
mẫu và bảo quản mẫu; Xử lý an toàn các dụng cụ đã được sử dụng để lấy mẫu; Các
mẫu bệnh phẩm nguy hiểm phải được đánh dấu xác nhận rõ ràng và được tiến hành
xét nghiệm cũng như xử lý chính xác; Các mẫu bị đổ hoặc các dụng cụ vỡ phải
được xử lý an toàn; Hạn chế những nguy cơ có thể xẩy ra để đảm bảo an toàn cho
người lấy mẫu, người vận chuyển mẫu, cho cộng đồng nói chung và cho phòng xét
nghiệm tiếp nhận mẫu nói riêng.
1.2.4. Vận chuyển mẫu xét nghiệm
Hệ thống vận chuyển mẫu xét nghiệm cần đảm bảo chính xác về mặt thời gian,
vị trí tiếp nhận mẫu và hạn chế tối đa những rủi ro đối với phòng xét nghiệm và
cả cộng đồng.
Các quy định về vận chuyển mẫu xét nghiệm phải phù hợp với các luật lệ đã
đề ra và đặc biệt tuân thủ theo quy chuẩn của tổ chức Y tế thế giới.
1.2.5. Nhận mẫu
Cần thu thập mẫu bệnh phẩm một cách hiệu quả và an toàn để xét nghiệm có
thể thực hiện đúng. Do vậy, cần có quy trình nhận mẫu bệnh phẩm bao gồm các
bước sau: Xác định mẫu bệnh phẩm có liên quan đến yêu cầu xét nghiệm; Ghi vào
sổ phiếu yêu cầu và các thông tin về mẫu bệnh phẩm; Ghi ngày và thời gian nhận;
Quy định về xử lý các mẫu bệnh phẩm cấp thiết. Trong trường hợp loại bỏ mẫu
bệnh phẩm không xét nghiệm, cần đưa ra các tiêu chuẩn loại bỏ, ghi chép lại các
mẫu bệnh phẩm đã loại bỏ.
2. Giai đoạn xét nghiệm
2.1. Các
yêu cầu cho giai đoạn xét nghiệm
Phòng xét nghiệm cần đảm bảo thực hiện xét nghiệm theo quy trình chuẩn thức
phù hợp, nhằm đạt được kết quả với độ chính xác cao, đáp ứng yêu cầu xét
nghiệm. Quy trình thực hiện xét nghiệm phải có tính chuẩn thức, được công nhận
và được cập nhật. Các quy trình phải được viết bằng ngôn ngữ dễ hiểu đối với
nhân viên, theo mẫu quy định và luôn được để sẵn trên bàn làm việc để tiện tham
khảo. Phòng xét nghiệm cần có danh sách các quy trình đang sử dụng, bao gồm yêu
cầu mẫu bệnh phẩm ban đầu sử dụng để xét nghiệm và các yêu cầu khác đối với nơi
yêu cầu xét nghiệm, các thông tin cần thiết để tham khảo. Phòng xét nghiệm phải
xây dựng kế hoạch và quy trình định kỳ rà soát, chỉnh sửa thay đổi, bổ sung nếu
cần để ngày càng hoàn thiện các quy trình kỹ thuật đảm bảo chất lượng xét
nghiệm. Các quy trình xét nghiệm phải được quản lý theo quy định chung của hệ
thống quản lý hồ sơ (Hồ sơ phương pháp).
2.2. Quá
trình thực hiện giai đoạn xét nghiệm
Việc lựa chọn các quy trình xét nghiệm cần rõ ràng, thích hợp và được đánh
giá thường xuyên bởi các chuyên gia trong lĩnh vực chuyên môn bao gồm: Các quy
trình lấy mẫu bệnh phẩm cần đạt yêu cầu để xét nghiệm và sự chấp nhận việc lấy
mẫu bệnh phẩm của người bệnh; Các quy trình xét nghiệm trước khi đưa vào sử
dụng, cần được ghi chép lại các phương pháp đã sử dụng và kết quả đạt được; Khi
thay đổi quy trình dẫn đến kết quả thu được có thể thay đổi, cần giải thích
trước khi đưa quy trình vào sử dụng.
Hầu hết các quy trình tại các phòng xét nghiệm chuẩn luôn được thẩm định
trước khi đưa vào sử dụng để đánh giá độ tin cậy của phương pháp. Đối với những
phương pháp thay đổi so với phương pháp gốc, luôn được thẩm định và so sánh độ
tin cậy với phương pháp tiêu chuẩn.
2.3. Bảo
đảm chất lượng xét nghiệm
Phòng xét nghiệm cần bảo đảm chất lượng xét nghiệm bằng cách thực hiện xét
nghiệm trong một điều kiện được kiểm soát, bao gồm: Việc triển khai các quy
trình tiền xét nghiệm một cách thích hợp; Các điều kiện về môi trường, thiết
bị, vật liệu và hệ thống thông tin, sử dụng các quy trình đã được chấp nhận; Có
sử dụng nội kiểm tra chất lượng để xác định những vấn đề sai sót có thể xảy ra;
Kiểm soát tính phù hợp của kết quả; Có tham gia các hệ thống ngoại kiểm tra để
duy trì và củng cố chất lượng xét nghiệm và có quy trình nội kiểm tra chất
lượng cho tất cả xét nghiệm đã được đánh giá đạt được chất lượng như yêu cầu.
2.3.1. Các sai số có thể gặp trong giai
đoạn làm xét nghiệm:
- Sai số ngẫu nhiên (Random errors): là sai số khó tránh, xảy ra một cách
ngẫu nhiên. Một số nguyên nhân có thể gây ra sai số này là:
+ Thuốc thử hỏng
+ Dụng cụ do thể tích không chính xác
+ Máy do và các thiết bị làm xét nghiệm không ổn định
+ Thao tác của nhân viên làm xét nghiệm chưa thuần thục.
- Sai số hệ thống (Systematic errors), đây là sai số thường gặp trong quá
trình làm xét nghiệm. Tuy nhiên, trên thực tế sai số này chủ yếu gặp do các
nguyên nhân sau đây:
+ Chất lượng thuốc thử không đảm bảo (không thực sự chuẩn)
+ Kỹ thuật xét nghiệm không đặc hiệu
+ Hóa chất chuẩn sai hoặc không chính xác
Để loại bỏ sai số này, chỉ có thể thực hiện được sau khi phát hiện được
nguyên nhân gây sai số.
- Sai số bất thường (Gross error): hay còn gọi là sai số “thô bạo”. Thông
thường sai số này xảy ra do nguyên nhân:
+ Không thực hiện đúng trình tự làm xét nghiệm
+ Nhầm thuốc thử, dụng cụ đo lường, nhầm bước sóng khi do màu
+ Tính kết quả sai.
Loại sai số này có thể tránh được nếu các xét nghiệm viên thận trọng trong
quá trình làm xét nghiệm và tổ chức tốt phòng xét nghiệm.
2.3.2. Các thông số đánh giá:
- Xét nghiệm định tính:
Tổng
số các kết quả dương tính
+ Độ
nhạy (Sensitivity): ----------------------------------------
Tổng
số các bệnh nhân bị bệnh
Độ nhạy của phương pháp xét nghiệm
càng lớn thì số lượng kết quả xét nghiệm âm tính giả càng thấp.
Ví dụ: Độ nhạy của thạch MacConkey trong phân lập Salmonella typhi thường kém. Vì vậy, vi khuẩn gây bệnh quan trọng
này có thể bị âm tính giả do sự phát triển mạnh mẽ của các vi khuẩn đường ruột
không gây bệnh.
+ Độ đặc hiệu (Specificity): ------------------------------------------------------
Tổng
số các bệnh nhân không bị bệnh
Độ đặc hiệu của phương pháp xét nghiệm càng lớn thì số lượng kết quả xét
nghiệm dương tính giả càng thấp.
Ví dụ: Độ đặc hiệu của phương pháp nhuộm Ziehl – Neelsen từ bệnh phẩm đờm
để chẩn đoán bệnh lao rất cao, do đó rất hiếm khi cho kết quả dương tính giả;
Độ đặc hiệu của phương pháp nhuộm Ziehl – Neelsen từ bệnh phẩm nước tiểu để
chẩn đoán bệnh lao thường thấp, bởi vì kỹ thuật này có thể cho kết quả dương
tính giả (như một số Mycobacterium không điển hình).
|
Độ nhạy và độ đặc hiệu của một xét nghiệm có liên
quan tương hỗ với nhau. Trong một số xét nghiệm, khi tăng độ nhạy của kỹ thuật
xét nghiệm thì lại dẫn đến giảm độ đặc hiệu, và ngược lại. Một xét nghiệm có độ
tin cậy cao khi độ nhạy và độ đặc hiệu cao.
+ Độ chính xác tương đối
(Relative accuracy), %:

Trong đó:
PA (Positive agreement): Xác suất chẩn đoán đúng ở những trường hợp cho kết
quả dương tính;
NA (Negative agreement): Xác suất chẩn đoán đúng ở những trường hợp cho kết
quả âm tính;
PD (Positive deviation): Xác suất chẩn đoán sai ở những trường hợp cho kết
quả dương tính;
ND (Negative deviation): Xác suất chẩn đoán sai ở những trường hợp cho kết
quả âm tính.
Tiêu chí đánh giá:
Thông thường các xét nghiệm được xác định là đạt tiêu khuẩn khi có độ nhạy, độ đặc hiệu và độ chính xác
tương đối đạt 95% trở lên. Tuy nhiên, tùy theo mục đích của mỗi loại xét
nghiệm mà yêu cầu về độ nhạy và độ đặc hiệu có thể thay đổi. Trong trường hợp
xét nghiệm dùng để sàng lọc thì độ nhạy cần quan tâm nhất, nhưng nếu những xét
nghiệm dùng trong chẩn đoán xác định thì độ đặc hiệu được quan tâm hơn.
- Xét nghiệm định lượng:
Độ đúng chỉ mức
độ gần nhau giữa giá trị trung bình của kết quả xét nghiệm và giá trị thực hoặc
giá trị được chấp nhận là đúng (μ). Độ chụm chỉ mức độ mức độ dao động của các kết
quả xét nghiệm độc lập quanh trị giá trung bình. Hình 1 mô tả sự mối quan hệ
giữa độ chụm, độ đúng và độ chính xác.
Độ chính xác
(accuracy) = độ chụm (precision) + độ đúng (trueness)
 |
| Hình 1: Minh họa khái niệm độ chính xác (độ chụm và độ đúng) |
Bảng 1: Sự khác nhau giữa độ lặp
lại, độ chụm trung gian và độ tái lập
|
Điều kiện
|
Độ lặp lại
|
Độ chụm trung gian
|
Độ tái lập
|
|
Nền mẫu
|
Khác
|
Khác
|
Khác
|
|
Nồng độ
|
Khác
|
Khác
|
Khác
|
|
Thiết bị
|
Giống
|
Khác
|
Khác
|
|
Người phân tích
|
Giống
|
Khác
|
Khác
|
|
Dụng cụ, hóa chất
|
Giống
|
Khác
|
Khác
|
|
Điều kiện môi trường
|
Giống
|
Khác
|
Khác
|
|
Phòng thử nghiệm
|
Giống
|
Giống
|
Khác
|
Cách xác định
Cách 1. Bố trí xét nghiệm
Tiến hành làm xét nghiệm lặp 10 lần
(ít nhất 6 lần) trên cùng một mẫu (mỗi lần bắt đầu từ cân hay đong mẫu). Mẫu
phân tích có thể là mẫu chuẩn, hoặc mẫu trắng có thêm chuẩn, tốt nhất là làm
trên mẫu thử hay mẫu thử thêm chuẩn.
Từng phòng xét nghiệm, có thể bố trí
xét nghiệm để tính độ lặp lại hoặc độ chụm trung gian. Trong một số trường hợp
tham gia so sánh với các phòng xét nghiệm khác (ví dụ trong chương trình thử
nghiệm thành thạo, đánh giá so sánh liên phòng)
Nên tiến hành ở các nồng độ mẫu khác
nhau (trung bình, thấp, cao) trong khoảng làm việc, mỗi nồng độ làm lặp lại 10
lần (ít nhất 6 lần). Tính độ lệch chuẩn (SD) và độ lệch chuẩn tương đối (RSD)
hay hệ số biến thiên (CV) theo các công thức sau:

Trong đó:
SD: độ lệch chuẩn
n: số lần thí nghiệm
xi: Giá trị tính được của lần thử
nghiệm thứ “i”
ẍ: Giá trị trung bình của các lần thử nghiệm
RSD%: Độ lệch chuẩn tương đối
CV%: Hệ số biến thiên
Cách 2. Tính toán trên các kết quả phân tích mẫu thực đã làm
Trong một số trường hợp việc ước
lượng độ lặp lại có thể thông qua tính toán dựa trên kết quả phân tích các mẫu
thực. Do đó việc lưu giữ các kết quả phân tích có vai trò rất quan trọng.
Dựa trên kết quả phân tích làm trên
mẫu thực trong nhiều tuần ít nhất là 10
mẫu, có thể là các nền mẫu khác nhau, nồng độ khác nhau nhưng phải có kết quả
làm lặp 2 lần .
Trường hợp các mẫu có nồng độ, hàm
lượng gần như nhau. Tính độ lệch giữa hai kết quả lặp của mỗi mẫu di
rồi tính độ lệch trung bình dtb, sau đó tính độ lệch chuẩn s:

Nếu các mẫu có nồng độ xi
khác nhau nhiều thì thay cho độ lệch di , tính độ lệch tương đối Di,
và độ lệch tương đối trung bình Dtb và sau đó tính độ lệch chuẩn
tương đối:

Tiêu chí đánh giá:
Đối chiếu trị giá tính được với trị
giá mong muốn hay giá trị yêu cầu hoặc so với RSD% lặp lại cho trong bảng 2
(RSD% tính được không được lớn hơn trị giá
trong bảng ở hàm lượng chất tương ứng). Độ chụm thay đổi theo nồng độ chất
phân tích. Nồng độ chất càng thấp thì kết quả càng dao động nhiều (không chụm)
nghĩa là RSD càng lớn.
Bảng 2: Độ lặp lại tối đa
chấp nhận tại các nồng độ khác nhau
(theo AOAC)
|
TT
|
Hàm lượng %
|
Tỷ lệ chất
|
Đơn vị
|
RSD (%)
|
|
1.
|
100
|
1
|
100%
|
1,3
|
|
2.
|
10
|
10-1
|
10%
|
1,8
|
|
3.
|
1
|
10-2
|
1%
|
2,7
|
|
4.
|
0,1
|
10-3
|
0,1 %
|
3,7
|
|
5.
|
0,01
|
10-4
|
100 ppm
|
5,3
|
|
6.
|
0,001
|
10-5
|
10 ppm
|
7,3
|
|
7.
|
0,0001
|
10-6
|
1 ppm
|
11
|
|
8.
|
0,00001
|
10-7
|
100 ppb
|
15
|
|
9.
|
0,000001
|
10-8
|
10 ppb
|
21
|
|
10.
|
0,0000001
|
10-9
|
1 ppb
|
30
|
Trong
nội bộ mỗi phòng thử nghiệm ngoài việc tính toán độ lặp lại, thì cần phải bố
trí xét nghiệm để tính được độ chụm trung gian (một số tác giả gọi là độ tái
lập nội bộ phòng thử nghiệm) theo một trong các cách sau đây:
+ Sử dụng các nhân viên thử
nghiệm khác nhau
+ Sử dụng các thiết bị với
một số đặc tính khác nhau, ví dụ các hệ thống HPLC khác nhau của các hãng khác
nhau, hoặc của cùng một hãng nhưng với các model khác nhau
+ Sử dụng các dung môi hóa
chất, thuốc thử có chất lượng khác nhau
+ Khác nhau về nhiệt độ và
độ ẩm của phòng thử nghiệm
+ Khác nhau các điều kiện cụ
thể của thiết bị, ví dụ thành phần dung môi pha động, tốc độ dòng, pH của pha
động,...
Việc tham gia thử nghiệm thành thạo, so sánh liên phòng
xét nghiệm là điều kiện rất quan trọng trong đánh giá phương pháp. Các phòng
xét nghiệm tham gia phải có kết quả xét nghiệm liên phòng trước khi được công
nhận đạt ISO.
+ Độ
chụm:
Trong nhiều trường hợp các xét nghiệm
trên những đối tượng và với những điều kiện khác nhau thường không cho kết quả
giống nhau. Điều này do các sai số ngẫu nhiên của mỗi quy trình gây ra và không
thể kiểm soát được hoàn toàn tất cả các yếu tố ảnh hưởng đến kết quả thử
nghiệm. Do đó, để kiểm soát được các sai số này, phải dùng đến khái niệm độ
chụm. Độ chụm chỉ phụ thuộc vào sai số ngẫu nhiên và không liên quan đến giá
trị thực. Độ chụm là một khái niệm định tính và được biểu thị định lượng bằng
độ lệch chuẩn hay hệ số biến thiên. Độ chụm càng thấp thì độ lệch chuẩn hay hệ
số biến thiên càng lớn.
Độ chụm có thể được phân ra thành 3
trường hợp sau:
● Độ lặp lại (Repeatability)
● Độ chụm trung gian (Intermediate
precision)
● Độ tái lập (Reproducibility)
Giá trị
trung bình (Mean):

Độ lệch chuẩn (Standard deviation):
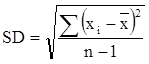
Độ đúng của phương pháp là khái niệm chỉ mức độ gần nhau
giữa giá trị trung bình của kết quả xét nghiệm và giá trị thực hoặc giá trị
được chấp nhận là đúng (μ).
Đối với đa số mẫu phân tích, giá trị
thực không thể biết một cách chính xác. Tuy nhiên, nó có thể có một giá trị quy
chiếu được chấp nhận là đúng (gọi chung là giá trị đúng).
Giống như độ chụm, độ đúng là một
khái niệm định tính. Độ đúng thường được diễn tả bằng độ chệch (bias).

Trong đó: Δ : Độ chệch (bias), %
Xtb: Giá trị trung
bình của kết quả thử nghiệm
μ: Giá trị thực hoặc giá
trị được chấp nhận là đúng
Cách xác định độ đúng:
Muốn xác định độ đúng cần phải tìm được giá trị
đúng, có nhiều cách khác nhau để xác định độ đúng, bao gồm việc so sánh kết quả
với kết quả thực hiện bởi một phương pháp đối chiếu hoặc sử dụng mẫu đã biết
nồng độ (mẫu kiểm tra hoặc mẫu chuẩn được chứng nhận) và phương pháp xác định
độ thu hồi (độ tìm lại).
Cách 1: So sánh với phương pháp chuẩn/đối chiếu
Phân tích mẫu chuẩn hoặc mẫu thử, thực hiện 10 lần
bằng phương pháp khảo sát và bằng một phương pháp đối chiếu. Phương pháp đối
chiếu tốt nhất là phương pháp tiêu chuẩn của các tổ chức có uy tín, nếu không
phương pháp đối chiếu là phương pháp đã qua thẩm định cho kết quả tin cậy trong
dải đo đang thực hiện. Tính toán các kết quả trung bình và độ lặp lại (hệ số
biến thiên) của hai phương pháp.
Đánh
giá độ tương đồng về độ chụm của 2 phương pháp bằng cách so sánh phương sai s2
của 2 phương pháp đó, dùng tiêu chuẩn F (Fisher) và so sánh hai trị giá trung
bình bằng tiêu chuẩn t (Student). Việc bố trí các thí
nghiệm phải được thực hiện theo phương pháp tham chiếu một cách nghiêm ngặt và
các phép đo phải được tiến hành dưới điều kiện lặp lại.
● So sánh hai phương sai (chuẩn F – Fisher)
Chuẩn F dùng để so sánh độ lặp lại của hai tập số
liệu hoặc hai phương pháp khác nhau. Với tập số liệu nhỏ, tính toán giá trị Ftn
(F thực nghiệm) theo công thức sau đây và so sánh với giá trị Fc (F
tra bảng)

Trong đó: Ftn: Giá trị F thực nghiệm
S12 , S22 : Các phương sai của hai phương pháp với quy ước S12 > S22 .
Nếu:
Ftn≤ Fc (α, k1, k2): Hai phương
pháp có độ lặp lại (độ chụm) giống nhau.
Nếu: Ftn > Fc (α,
k1, k2): Hai phương pháp có độ lặp lại khác nhau, trong
trường hợp này nếu độ lặp lại của phương pháp thử nghiệm khác với phương pháp
chuẩn thì cần xem xét thêm về độ lặp lại như đã mô tả trong phần trên.
Trong đó: Fc (α, k1,
k2): Giá trị F tra bảng (xem phụ lục) với:
k1, k2 : Bậc tự do (k1
= n1-1; k2 = n2-1)
n1,
n2: Số lần làm thực nghiệm của hai phương pháp
α:
Mức ý nghĩa (significance level), thường lấy α = 0,05 (tương ứng với độ tin cậy
(confidence level) 95%)
● So sánh hai giá trị trung bình
(chuẩn t – Student)
Chuẩn t
được dùng để so sánh xem có sự khác nhau giữa giá trị thực nghiệm và giá trị
thực hay không; phương pháp này được ứng dụng hoặc để so sánh kết quả thực
nghiệm với giá trị chuẩn trong mẫu kiểm tra (xem thêm cách 2) hoặc để so sánh
kết quả của phương pháp phân tích với phương pháp đối chiếu.
Trước khi so sánh hai giá
trị trung bình cần so sánh hai phương sai. Với số lần phân tích nhỏ hơn 30, khi
hai phương sai có sự đồng nhất, tính độ lệch chuẩn chung và giá trị ttn
(t thực nghiệm) theo công thức sau
đây và so sánh với giá trị tc(t tra bảng):


k
= n1+n2-2
Trong
đó:
ttn: Giá trị t thực nghiệm
tc(α, k): Giá
trị t tra bảng mức ý nghĩa α, bậc tự do k (xem phụ lục 1)
n1, n2 : Số lần thí nghiệm lần lượt của phương pháp
thử và phương pháp đối chiếu
S12 , S22 : Phương sai lần lượt
của phương pháp thử nghiệm và của phương pháp đối chiếu
 : Giá trị trung bình
lần lượt của phương pháp thử nghiệm và của phương pháp đối chiếu
: Giá trị trung bình
lần lượt của phương pháp thử nghiệm và của phương pháp đối chiếu
Nếu
ttn ≤ tc(α, k): Không có sự khác nhau về kết quả của hai phương pháp.
Nếu
ttn > tc(α, k): Có sự khác nhau về kết quả của hai phương pháp, phương pháp thử
nghiệm mắc sai số hệ thống.
Trong trường hợp hai
phương sai không đồng nhất (khác nhau có ý nghĩa), tính giá trị ttn
và bậc tự do k theo các công thức sau và so sánh như trên.


Cách 2: Sử dụng vật liệu chuẩn (Reference material)
Vật liệu chuẩn (còn gọi là mẫu chuẩn) là mẫu phân
tích có hàm lượng chất phân tích đã được xác định trước và là đúng. Có nhiều
cấp vật liệu chuẩn khác nhau, trong đó cao nhất là CRM (certified reference
material – vật liệu chuẩn được chứng nhận) được cung cấp bởi các tổ chức có uy
tín trên thế giới.
Các mẫu CRM luôn có kết quả kèm theo khoảng dao
động, do đó khi phân tích mẫu CRM có thể đánh giá được độ đúng dựa vào khoảng
dao động cho phép (ví dụ: Mẫu thịt, chỉ tiêu clenbuterol là 1 ng/g ± 0,05 ng/g;
nếu kết quả phân tích được trong khoảng từ 0,95-1,05 thì đạt).
Nếu không có các mẫu CRM có thể sử dụng các mẫu kiểm
tra (QC-Quality Control) đã biết nồng độ. Phòng thử nghiệm có thể tự chuẩn bị
các loại mẫu này, hoặc sử dụng các mẫu thực có hàm lượng đã biết hoặc sử dụng
các mẫu lưu từ chương trình so sánh liên phòng thử nghiệm. Trong trường hợp
khác phòng thử nghiệm có thể sử dụng các mẫu thêm chuẩn để đánh giá độ đúng,
nội dung này sẽ được mô tả cụ thể trong cách 3 dưới đây.
Nhiều tổ chức có uy tín như USFDA, EURACHEM, ICH ...
quy định tính độ chệch (bias) để xác định độ đúng như sau:

USFDA quy định độ chệch của các phương pháp xác định
dư lượng phải không được lớn hơn 15% và không lớn hơn 20% tại LOQ.
Có thể sử dụng chuẩn t để đánh giá kết quả
như sau:
Phân tích mẫu chuẩn lặp lại 10
lần (tối thiểu 6 lần), tính giá trị trung bình và độ lệch chuẩn, từ đó tính giá
trị ttn theo công thức sau đây và so sánh với tc (p):

Nếu ttn ≤ tc
: Không có sự khác nhau về kết quả của giá trị trung bình so với giá trị tham
chiếu ở mức ý nghĩa α, tức là phương pháp có độ đúng đạt yêu cầu.
Nếu ttn > tc:
Có sự khác nhau về kết quả của phương pháp thử nghiệm so với kết quả tham chiếu
ở mức ý nghĩa α, phương pháp thử nghiệm mắc sai số hệ thống.
Cách 3: Xác định độ thu hồi
Các phương pháp tính độ đúng theo cách 1 hay cách 2
đều gặp những khó khăn nhất định. Trong nhiều trường hợp không thể tìm hoặc áp
dụng một phương pháp tiêu chuẩn để so sánh kết quả, cũng như không thể dễ dàng
có được các mẫu chuẩn hoặc mẫu chuẩn được chứng nhận phù hợp với phương pháp.
Việc xác định độ đúng do đó có thể thực hiện thông qua xác định độ thu hồi (còn
gọi là độ tìm lại) của phương pháp.
Thêm một lượng chất chuẩn xác định vào mẫu thử hoặc
mẫu trắng, phân tích các mẫu thêm chuẩn đó, làm lặp lại tối thiểu 4 lần bằng
phương pháp khảo sát, tính độ thu hồi theo công thức sau đây:
+
Đối với mẫu thử:

+
Đối với mẫu trắng:

Trong đó: R%: Độ
thu hồi, %
Cm+c:
Nồng độ chất phân tích trong mẫu thêm chuẩn
Cm:
Nồng độ chất phân tích trong mẫu thử
Cc:
Nồng độ chuẩn thêm (lý thuyết)
Ctt:
Nồng độ chất phân tích trong mẫu trắng thêm chuẩn
Sau đó
tính độ thu hồi chung là trung bình của độ thu hồi các lần làm lặp lại.
Thêm chất chuẩn ở 3 mức nồng độ là mức thấp, trung
bình và cao trong khoảng nồng độ làm việc. Theo quy định của hội đồng châu Âu
đối với các chỉ tiêu an toàn (các chỉ tiêu thuộc nhóm độc, có quy định giới hạn
cho phép, ví dụ tồn dư hocmon, kháng sinh, hóa chất bảo vệ thực vật...) thêm
chuẩn vào mẫu trắng ở 3 mức nồng độ tại 0,5 lần, 1 lần và 2 lần giới hạn cho
phép (MRL).
Sau khi đánh giá độ thu hồi, so sánh kết quả với các
giá trị cho bởi trong bảng 3. Độ thu hồi ở các nồng độ khác nhau có kỳ vọng
khác nhau. Trong trường hợp phân tích các chất hàm lượng vết có thể tham khảo
tiêu chuẩn của hội đồng châu Âu (bảng 4)
Bảng 3: Độ thu hồi chấp
nhận ở các nồng độ khác nhau (theo AOAC)
|
TT
|
Hàm lượng [%]
|
Tỷ lệ chất
|
Đơn vị
|
Độ thu hồi [%]
|
|
1.
|
100
|
1
|
100%
|
98-102
|
|
2.
|
>=10
|
10-1
|
10%
|
98-102
|
|
3.
|
>=1
|
10-2
|
1%
|
97-103
|
|
4.
|
>=0,1
|
10-3
|
0,1 %
|
95-105
|
|
5.
|
0,01
|
10-4
|
100 ppm
|
90-107
|
|
6.
|
0,001
|
10-5
|
10 ppm
|
80-110
|
|
7.
|
0,0001
|
10-6
|
1 ppm
|
80-110
|
|
8.
|
0,00001
|
10-7
|
100 ppb
|
80-110
|
|
9.
|
0,000001
|
10-8
|
10 ppb
|
60-115
|
|
10.
|
0,0000001
|
10-9
|
1 ppb
|
40-120
|
Bảng 4: Quy định về độ thu hồi của hội đồng châu
Âu
|
TT
|
Hàm lượng chất
|
Đơn vị
|
Độ thu hồi [%]
|
|
1.
|
≤ 1 μg/kg
|
≤ 1 ppb
|
50%-120%
|
|
2.
|
> 1 μg/kg đến < 10
μg/kg
|
1-10 ppb
|
70%-110%
|
|
3.
|
³ 10 μg/kg
|
³ 10ppb
|
80%-110%
|
3. Giai đoạn sau xét nghiệm
3.1. Yêu
cầu của giai đoạn sau xét nghiệm
Sau khi đã hoàn thành xét nghiệm, việc đánh giá và ghi nhận kết quả, gửi
trả kết quả đến nơi yêu cầu xét nghiệm cần tránh
sai sót và chậm trễ, gây tổn hại đến uy tín của phòng xét nghiệm. Trả lời kết
quả xét nghiệm cần đảm bảo đủ thông tin và được lưu trữ ở phòng xét nghiệm để
tiện tham khảo hoặc tra cứu khi cần thiết.
3.2.
Thực hiện báo cáo kết quả của giai đoạn sau xét nghiệm
Mục tiêu của phòng xét nghiệm là ghi nhận kết quả xét nghiệm vào phiếu trả
lời kết quả xét nghiệm chính xác và đúng thời gian theo thời hạn đã xác định để
bảo đảm hiệu quả của phòng xét nghiệm đối với nơi yêu cầu xét nghiệm. Việc trả
lời kết quả có thể bằng phiếu trả lời kết quả; Trả lời qua diện thoại.
3.2.1. Báo cáo bằng văn bản
Phương pháp cơ bản để chuyển giao kết quả cho người yêu cầu xét nghiệm là
viết phiếu trả lời kết quả. Phiếu trả
lời kết quả phải rõ ràng, không mập mờ, chứa đựng đầy đủ các thông tin để người
yêu cầu xét nghiệm có thể hiểu được kết quả xét nghiệm. Phiếu trả lời kết quả cần
đưa các thông tin sau như: Tên phòng xét nghiệm; Thông tin về bệnh nhân; Tên
cơ sở (người) yêu cầu xét nghiệm/hoặc địa chỉ để gửi phiếu trả lời kết quả; Loại mẫu bệnh phẩm, ngày và giờ thu
thập mẫu bệnh phẩm; Thời gian và ngày
viết phiếu trả lời kết quả; Kết quả,
bao gồm nguyên nhân không xét nghiệm (nếu có).
3.2.2. Báo cáo qua điện thoại
Phòng xét nghiệm thường được yêu cầu thông báo kết quả qua điện thoại cho
người yêu cầu xét nghiệm. Phương pháp này cần được thực hiện rõ ràng nhằm giảm
thiểu nguy cơ sai sót. Nếu trả lời kết quả qua điện thoại, cần ghi chép lại các
thông tin sau: Tình huống thông báo kết quả; Giới thiệu tên người thông báo kết
quả; Tên người cần nhận kết quả; Cách xác định bệnh nhân giữa người thông báo
và người nhận kết quả; Có bằng chứng khẳng định kết quả đã được thông báo cho
đúng người, đúng nơi,
3.2.3. Thay đổi phiếu trả lời kết quả
Việc thay đổi phiếu trả lời kết quả được yêu cầu đối với phòng xét nghiệm
để thực hiện khi cần thiết. Tuy nhiên, cần tuân thủ các bước sau: Có tiêu chuẩn
để thực hiện việc thay đổi phiếu trả lời kết quả; Có nhân viên chịu trách nhiệm
thực hiện thay đổi phiếu trả lời kết quả; Thông báo cho người yêu cầu xét
nghiệm biết về sự thay đổi phiếu trả lời kết quả; Ghi chép lại việc thay đổi
phiếu trả lời kết quả; Nguyên nhân cần thay đổi.
3.3. Tư
vấn lâm sàng và giải nghĩa
Việc giải nghĩa kết quả xét nghiệm phải đáp ứng được nhu cầu và yêu cầu của
người yêu cầu xét nghiệm. Giải nghĩa ghi trên phiếu kết quả cần rõ ràng, súc
tích và không mập mờ. Chỉ những người đã được đào tạo và có thẩm quyền mới được
tư vấn lâm sàng và đưa ra những nhận xét giải nghĩa kết quả xét nghiệm.
TÀI
LIỆU THAM KHẢO
1.
International standard ISO 15189. 2007 (E). Published in
Switzerland.
2.
Laboratory Biosafety manual, Third edition. World Health
organization 2004.
3.
Clinical Pathology Accreditation (UK) Ltd, Standard for the
medical Laboratory. CPA, Version 2, September 2007.
4.
TCVN ISO/IEC 17025:2005, Yêu
cầu chung về năng lực của phòng thử nghiệm và hiệu chuẩn.
5.
AOAC International (2007), How to meet ISO 17025 requirements for method verification, USA.
6.
ICH (1996), Validation
of Analytical Procedures: Text and methodology, ICH Hamonised Tripartite
Guideline.
Phụ lục 1: Bảng
phân phối chuẩn Student với các mức ý nghĩa từ 0,10 đến 0,001
|
Bậc tự do
|
α
|
||||
|
0,10
|
0,05
|
0,01
|
0,005
|
0,001
|
|
|
1
|
6,314
|
12,71
|
31,82
|
63,66
|
318,3
|
|
2
|
2,920
|
4,303
|
6,965
|
9,925
|
22,33
|
|
3
|
2,353
|
3,182
|
4,541
|
5,841
|
10,21
|
|
4
|
2,132
|
2,776
|
3,747
|
4,604
|
7,173
|
|
5
|
2,015
|
2,571
|
3,365
|
4,032
|
5,893
|
|
6
|
1,943
|
2,447
|
3,143
|
3,707
|
5,208
|
|
7
|
1,895
|
2,365
|
2,998
|
3,499
|
4,785
|
|
8
|
1,860
|
2,306
|
2,896
|
3,355
|
4,501
|
|
9
|
1,833
|
2,262
|
2,821
|
3,250
|
4,297
|
|
10
|
1,812
|
2,228
|
2,764
|
3,169
|
4,144
|
|
11
|
1,796
|
2,201
|
2,718
|
3,106
|
4,025
|
|
12
|
1,782
|
2,179
|
2,681
|
3,055
|
3,930
|
|
13
|
1,771
|
2,160
|
2,650
|
3,012
|
3,852
|
|
14
|
1,761
|
2,145
|
2,624
|
2,977
|
3,787
|
|
15
|
1,753
|
2,131
|
2,602
|
2,947
|
3,733
|
|
16
|
1,746
|
2,120
|
2,583
|
2,921
|
3,686
|
|
17
|
1,740
|
2,110
|
2,567
|
2,898
|
3,646
|
|
18
|
1,734
|
2,101
|
2,552
|
2,878
|
3,610
|
|
19
|
1,729
|
2,093
|
2,539
|
2,861
|
3,579
|
|
20
|
1,725
|
2,086
|
2,528
|
2,845
|
3,552
|
|
21
|
1,721
|
2,080
|
2,518
|
2,831
|
3,527
|
|
22
|
1,717
|
2,074
|
2,508
|
2,819
|
3,505
|
|
23
|
1,714
|
2,069
|
2,500
|
2,807
|
3,485
|
|
24
|
1,711
|
2,064
|
2,492
|
2,797
|
3,467
|
|
25
|
1,708
|
2,060
|
2,485
|
2,787
|
3,450
|
|
26
|
1,706
|
2,056
|
2,479
|
2,779
|
3,435
|
|
27
|
1,703
|
2,052
|
2,473
|
2,771
|
3,421
|
|
28
|
1,701
|
2,048
|
2,467
|
2,763
|
3,408
|
|
29
|
1,699
|
2,045
|
2,462
|
2,756
|
3,396
|
|
30
|
1,697
|
2,042
|
2,457
|
2,750
|
3,385
|
|
|
1,645
|
1,960
|
2,326
|
2,576
|
3,090
|

|
Phụ lục 2 (tiếp theo)

No comments:
Post a Comment